Introduction
History
Causes and Symptoms
Epidemiology
Diagnosis and Treatment
References
Carnosinemia, also known as carnosinase deficiency, is an extremely rare inherited metabolic disorder. It presents with increased carnosine levels in urine and low levels or absence of carnosinase in the blood. It is associated with severe neurological symptoms in humans and is characterized by developmental delays and seizures. Common symptoms include tremors, psychomotor impairment, hypotonia, myoclonic seizures, and failure to form social relationships.
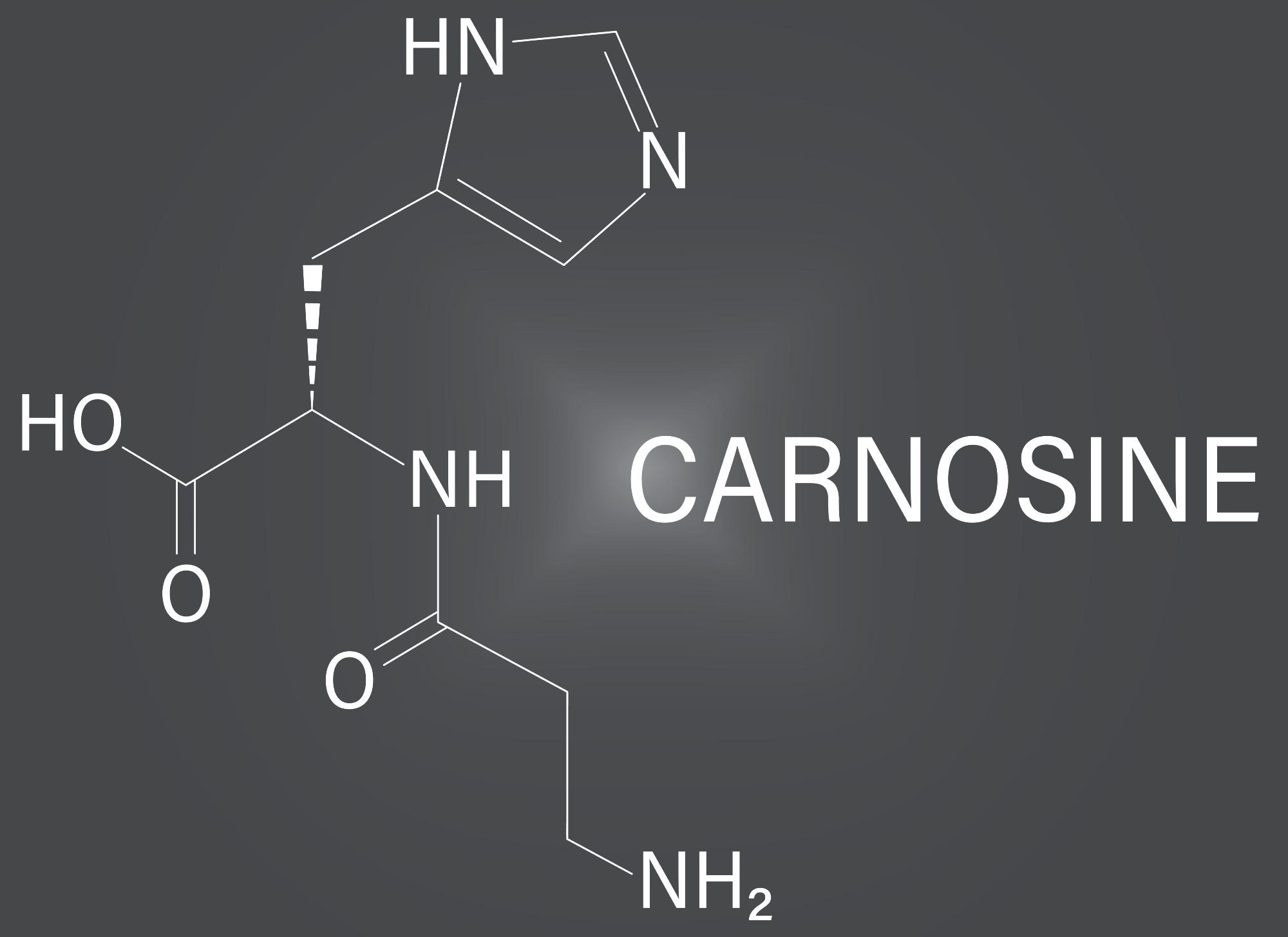
Carnosine is a dipeptide made up of beta-alanine and histidine found in skeletal muscle and nervous system cells. As a result of this condition, there is an accumulation of carnosine in the urine, cerebrospinal fluid, blood, and nerve tissue.
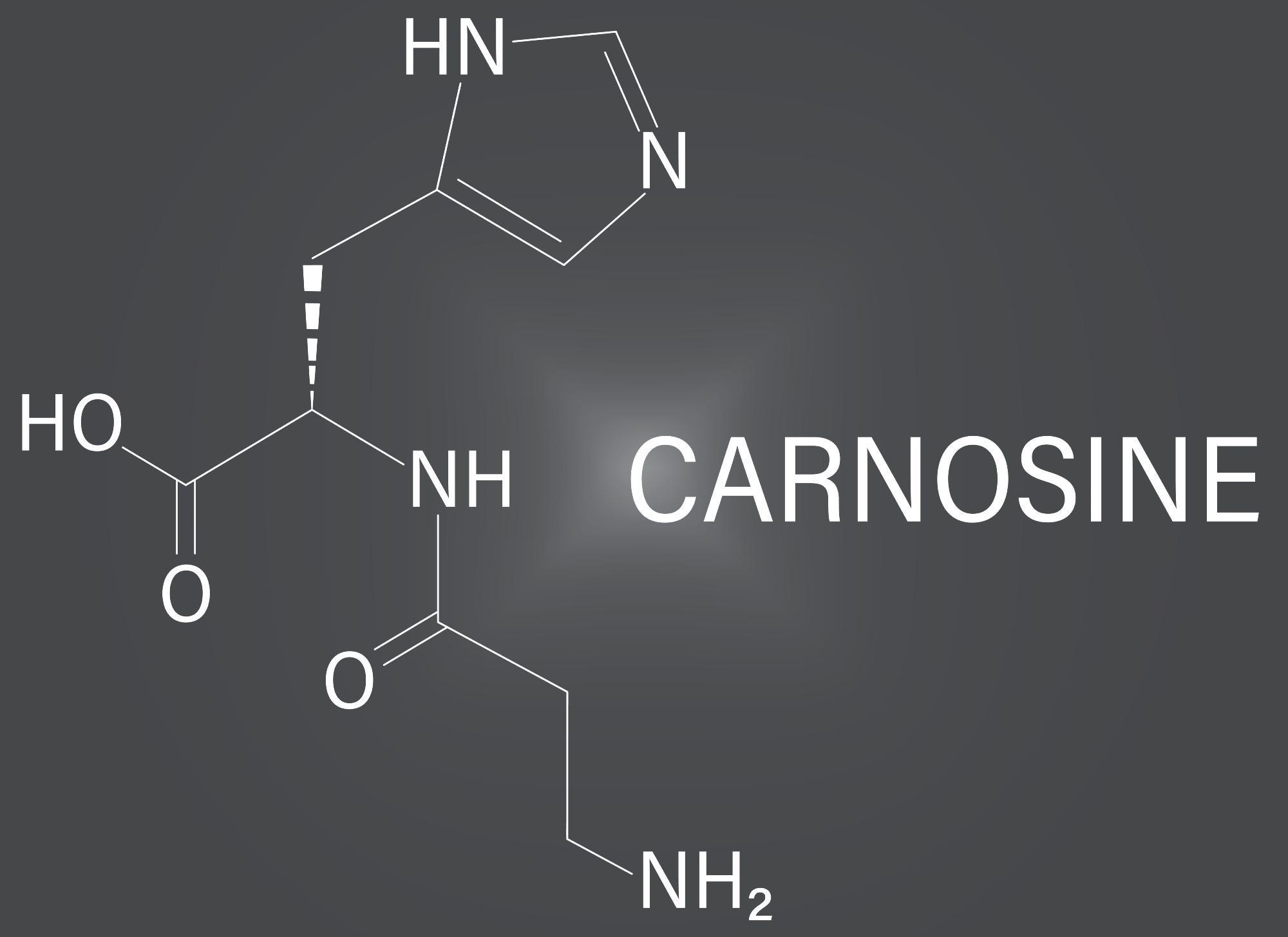
Image Credit: Spunt/Shutterstock.com
History
Carnosinaemia was initially characterized as an inherited metabolic disorder in 1967, and it was later shown to be caused by a lack of serum carnosinase in 1968. Perry et al. (1967) described two unrelated children who had a developing neurologic disorder with severe mental deficiency and myoclonic seizures. Even after all sources of the dipeptide were removed from the diet, both excreted carnosine in the urine.
Perry et al. (1967) speculated that one, or maybe both, had a carnosinase activity deficiency. One child was born to first-cousin parents and had German and Dutch ancestry. The other youngster came from a Chinese family. The enzyme in normal human serum that hydrolyzes the dipeptides carnosine and anserine into their constituent amino acids was nearly absent in the two patients, according to Perry et al. (1968).
Terplan and Cares (1972) documented another family with this unusual abnormality. Two brothers, aged seven and four, had passed away. A 6-year-old sister appeared to be normal, but she was experiencing chemical alterations. Carnosinase activity in the parents is low. The older boy's autopsy revealed extensive axonal degeneration, many spheroids in the grey matter, demyelination, fibrosis, and Purkinje fiber loss.
Willi et al. (1997) described a 30-month-old child with hypotonia, developmental delay, and tremor who excreted substantial levels of carnosine and anserine despite eating only small amounts of meat. These anomalies were improved but not eliminated by a strict meat-free diet. The activity of serum carnosinase was determined to be exceedingly low.
Causes and Symptoms
Although the specific nature of the biochemical anomaly that causes carnosinemia is unknown, carnosinase, the enzyme that breaks down carnosine, is present in both the brain and the blood. Studies of muscle tissue from affected people reveal that the enzyme carnosinase is aberrant in its metabolism of two dipeptides found in meats, carnosine and anserine.
The CNDP1 (Carnosine Dipeptidase 1) gene encodes carnosinase (CN1), an enzyme that degrades histidine-containing dipeptides like carnosine, anserine, and homocarnosine. Carnosinemia is assumed to be passed down through the family in an autosomal recessive fashion. The genetic basis of carnosinaemia has not been formally confirmed to be attributable to CNDP1 mutations, but reduced CN1 function should raise serum carnosine concentrations.
Carnosinaemia and serum carnosinase insufficiency have a wide range of clinical manifestations. 80-99% of patients experience a high level of carnosine in urine, loss of developmental milestones, and intellectual disability. Extreme drowsiness and seizures are common signs of carnosinemia in children under one year. Children with this condition have slowed growth, low muscle tone, motor difficulties, and intellectual developmental delays.
Myoclonic seizures can occur with seizures. Affected children show varying degrees of intellectual deficit by two, leading to intellectual impairments and developmental regression. Some of the impacted youngsters also have muscle weakness (congenital myopathy). A few patients with few to no symptoms have been diagnosed with this illness.
Carnosineuria (excess carnosine in the urine) and low levels of the enzyme carnosinase in the blood are common among affected persons. It's also unknown how low carnosinase and/or high carnosine levels in the body are linked to the disorder's neurological symptoms.
Epidemiology
A total of 30 people have been identified globally. Both males and females are at risk.
Diagnosis and Treatment
Testing the amounts of amino acids in the blood and/or urine can indicate abnormally high levels of carnosine and anserine in the serum and urine, indicating carnosinemia. The enzyme carnosinase in the blood can be detected via highly specialized blood tests. After eliminating meat from the diet, an amino acid study of serum and/or urine is used to make the diagnosis.
Because there is no effective medication treatment for carnosinemia, it is treated symptomatically and supportively. Carnosine, a dipeptide containing histidine and alanine, is abundant in meat. When carnosinase is deficient, carnosine levels rise in the blood and urine.
A strict meat-free diet can help with symptoms, but it will not cure the disease. A vegetarian diet lowers serum carnosine levels by reducing the amount of carnosine and anserine supplied to the body. However, it's uncertain whether this will have any impact on symptoms or disease progression. Carnosinemia patients and their families should seek genetic counseling.
References:
- Shivam Choudghal, Dr.C.Revathi M.Sc.(N), Ph.D.(N), Dr.B.Senthilkumar, Henrita Boro, Saksham Kumar, Drishti Sharma, & Harleen Kaur. (2021). A PERSPECTIVE REVIEW ON CARNOSINEMIA. International Journal of Allied Medical Sciences and Clinical Research, 9(3), 493-496. Retrieved from https://ijamscr.com/ijamscr/article/view/1054
- Caruso, G., Caraci, F., & Jolivet, R. B. (2019). Pivotal role of carnosine in the modulation of brain cells activity: Multimodal mechanism of action and therapeutic potential in neurodegenerative disorders. Progress in neurobiology, 175, 35–53. https://doi.org/10.1016/j.pneurobio.2018.12.004
- Peters, V., Zschocke, J., & Schmitt, C. P. (2018). Carnosinase, diabetes mellitus and the potential relevance of carnosinase deficiency. Journal of inherited metabolic disease, 41(1), 39–47. https://doi.org/10.1007/s10545-017-0099-2
- Carnosinemia. [Online] OMIM. Available at: https://omim.org/entry/212200
- Carnosinemia. [Online] NIH-GARD. Available at: https://rarediseases.info.nih.gov/diseases/6001/carnosinemia
- Carnosinemia. [Online] National Organization for Rare Disorders. Available at: https://rarediseases.org/rare-diseases/carnosinemia/
Further Reading
- All Rare Disease Content
- What is a Rare Disease?
- Teaching old drugs new tricks – drug repurposing for rare diseases
- What is Agnosia?
- What is Ameloblastoma?
Last Updated: Jun 1, 2022
.png)
Written by
Emily Henderson
During her time at AZoNetwork, Emily has interviewed over 300 leading experts in all areas of science and healthcare including the World Health Organization and the United Nations. She loves being at the forefront of exciting new research and sharing science stories with thought leaders all over the world.
Source: Read Full Article