
 and tubulin (red) distribution in young mouse cortical neurons deficient in giant ankyrin-B. Credit: University of North Carolina at Chapel Hill")
Scientists at the University of North Carolina at Chapel Hill School of Medicine and colleagues have demonstrated that rare variants in the ANK2 gene, consistently found in individuals with autism spectrum disorder (ASD), can alter architecture and organization of neurons, potentially contributing to autism and neurodevelopmental comorbidities.
The discovery, published in the journal eLife, was led by Damaris Lorenzo, Ph.D., an assistant professor in the UNC Department of Cell Biology and Physiology and member of the UNC Neuroscience Center and the UNC Intellectual and Developmental Disabilities Research Center.
ANK2 instructs neurons and other cell types how to make ankyrin-B, a protein with multiple functions in the nervous system. ANK2 encodes for various versions (isoforms) of ankyrin-B through a process known as alternatively splicing, whereby portions of the protein are excluded in the final molecules.
Mammals, such as mice and humans, express the full-size (giant) ankyrin-B isoform only in neurons; another highly abundant isoform half its size is found in virtually every type of cell and organ. Multiple genetic studies have consistently identified rare variants in ANK2 in individuals with ASD, making it one of the high-confidence risk genes associated with the condition. Depending on their type and location in the gene, ANK2 variants can affect giant ankyrin-B exclusively or both isoforms simultaneously.
“Together with its high prevalence and striking clinical presentation, ASD’s uncertain cause is a major limiting step in advancing therapeutic options,” Lorenzo said. “The evidence of ASD’s genetic origin is strong but also complex, with at least 100 other high-risk genes linked to the disorder.”
Revealing the cause of ASD is made even more complicated because single genes such as ANK2 and the isoforms they encode can have multiple cellular functions. However, based on how subgroups of these genes overlap functionally, or work together to enact biological pathways, scientists have proposed convergent mechanisms that may be predominantly affected in individuals with ASD. One of these common mechanisms is neuron communication, which is determined in part by alterations in axons—the long extensions that carry signals from neurons to other neurons. Underlying these processes within a single neuron is the axonal cytoskeleton, a complex network of filament-like proteins that plays pivotal roles in each neuron’s growth, shape, and plasticity. The axonal cytoskeleton is thought to be another major functional axis affected in ASD.
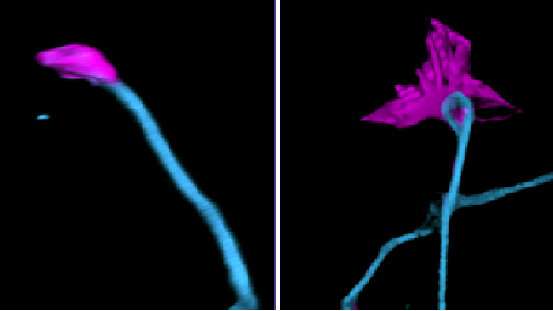
Previous work by Lorenzo published in JCB showed that simultaneous loss of both major isoforms of ankyrin-B in the brain of mice resulted in profound anatomical defects involving axonal wiring, underscoring the importance of ankyrin-B in brain architecture and function. In a later study published in PNAS, Lorenzo and colleagues from Duke University observed that eliminating only the giant ankyrin-B isoform in neurons cultured in the lab resulted in more axon branches, which implicated deficits in the dynamics of microtubules, an essential cytoskeleton component.
In this new study, the Lorenzo lab showed that selective loss of the giant ankyrin-B isoform leads to more axon branches in mouse brains and volumetric increases of multiple axonal bundles including the corpus callosum. In collaboration with Eva Anton, Ph.D., at the UNC Neuroscience Center and co-author of the ife paper, they found that the giant ankyrin-B isoform is required to maintain the topographic order of callosal axons arising from the somatosensory cortex during brain development and to ensure the specific targeting and refinement of callosal projections on the opposite side of the brain. The team did not observe increases in axon branches in a novel mouse model they engineered that only lacks the shorter ankyrin-B isoform.
“These findings confirm divergent roles of ankyrin-B isoforms and support critical and specialized roles of giant ankyrin-B in axon collateral branch formation, targeting, and refinement,” Lorenzo said.
In collaboration with a team led by Meng Meng Fu, Ph.D., at the National Institute of Neurological Disorders and Stroke and co-authors of the eLife paper, the researchers verified that the observed corpus callosum abnormalities did not involve changes in myelination and in the number and maturation of oligodendrocytes, a non-neuronal brain cell type implicated in similar pathologies.
“Brain cortical regions have been the most directly linked to ASD pathology. The changes we observed in cortical structural connectivity likely result from combined defects in axon branch initiation, guidance, and pruning of misdirected or overabundant projections during development due to giant ankyrin-B deficiency,” Lorenzo said.
Cues outside of cells modulate these processes to trigger changes in neurons through attractive and repulsive effects. Lorenzo’s research team showed that cortical neurons require the giant ankyrin-B isoform to make possible the repulsive effects of semaphorin 3A, a molecule that interacts with and collapses the tips of axons and their branches. The team also showed that ANK2 variants exclusively affecting giant ankyrin-B have similar loss of response to the Semaphorin 3A molecule, revealing a possible mechanistic contribution to ASD.
Source: Read Full Article